From Sequences to Community Composition¶
Data Source: https://github.com/awbrooks19/vmi_microbiome_bootcamp
What to do when you get back your sequence files?
Sequence File Formats¶
What do sequence file formats look like?
They can be displayed in a couple ways.
The first is as two separate files for the sequences and the quality scores
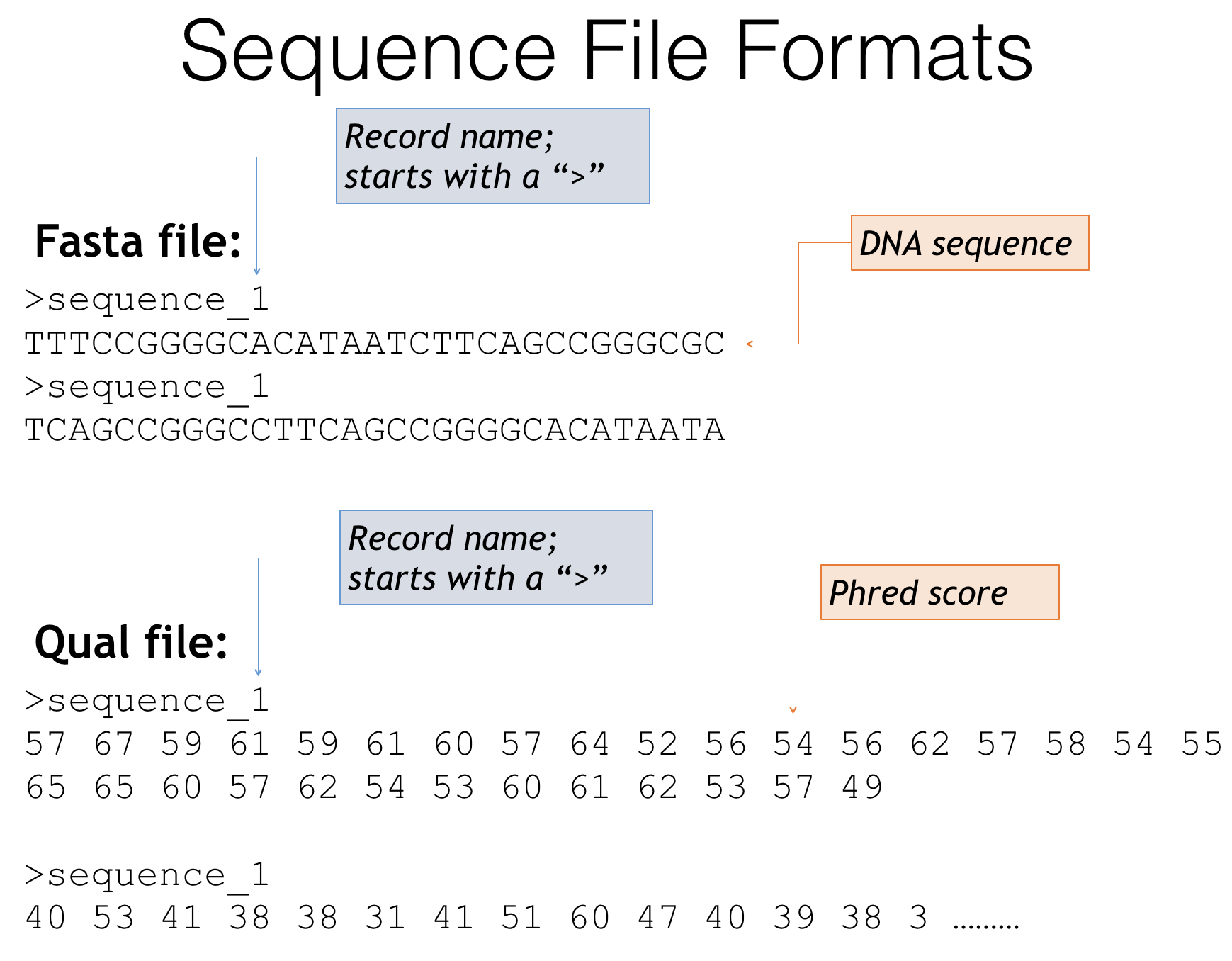
Fasta and quality score file formats
Fasta files - The genetic sequences of your amplicons
- Header lines always starts with a ‘>’
- Describes the sequence, often an id or incremental number
- Next line is the genetic sequence of your amplicon itself (A,T,C,G’s)
- Quality score files
- Start with the same headers as the fasta file
- Contain scores that describe how confident the sequencer is in that particular base call
- Often these are expressed as Phred scores:
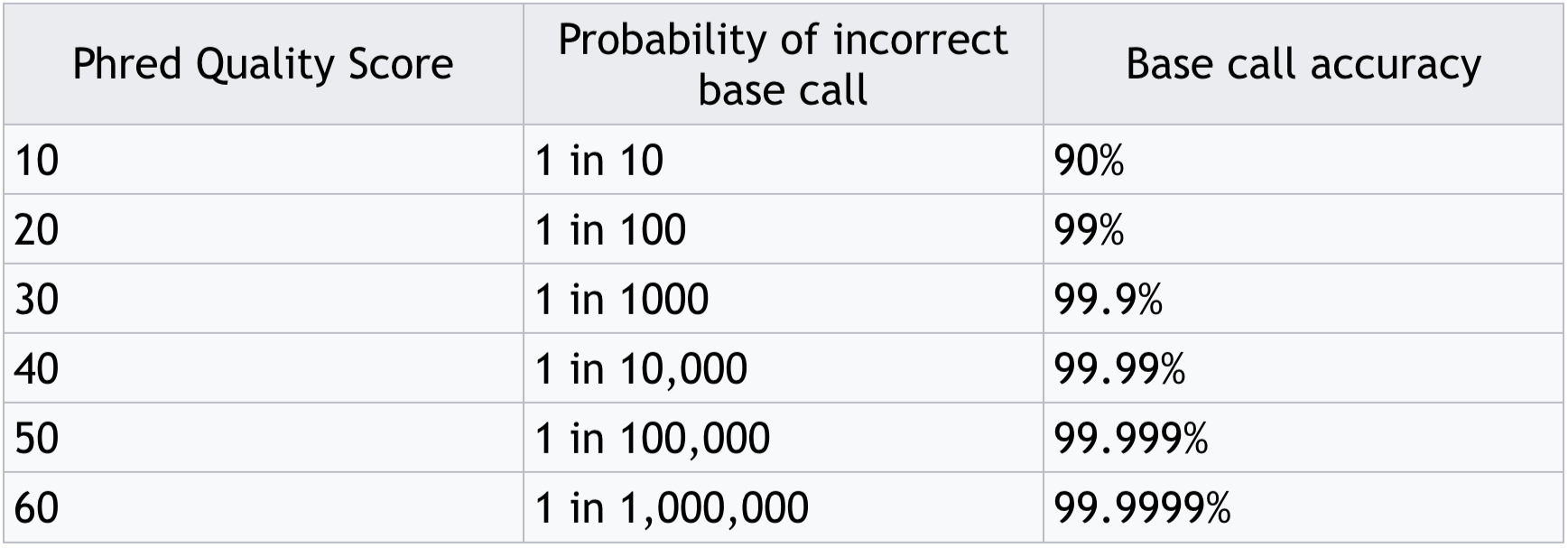
Phred score translation to base call accuracy
A second way you may find you files is a fastq file
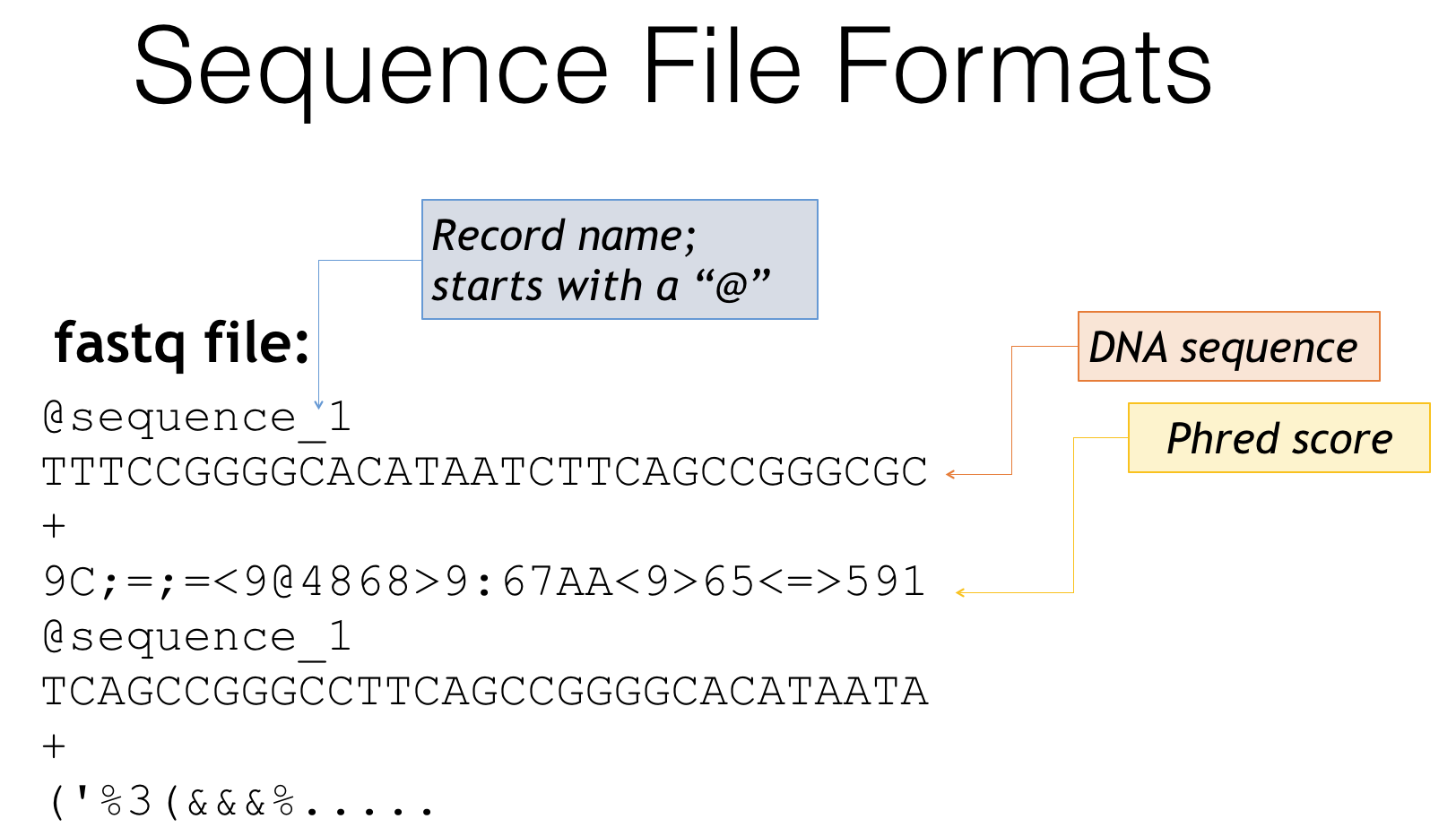
Fastq file format
- Fastq is essentially a combination of fasta and quality score files
- Header lines often start with ‘@’
- Followed by the genetic sequence like a fasta file
- The third line is generally just a ‘+’
- The fourth line is a condensed form of the quality scores
- Then the pattern repeats for the next sequence
Some sequencing centers perform some quality processing, others don’t. Make sure you ask what has been done!
- If they try to provide another format or raw data, ask for one of these two options. This is standard.
- Always get quality scores, even if they “did quality control for you”
Types of Sequences in QIIME2¶
How do you make QIIME2 aware of your sequences and mapping file?
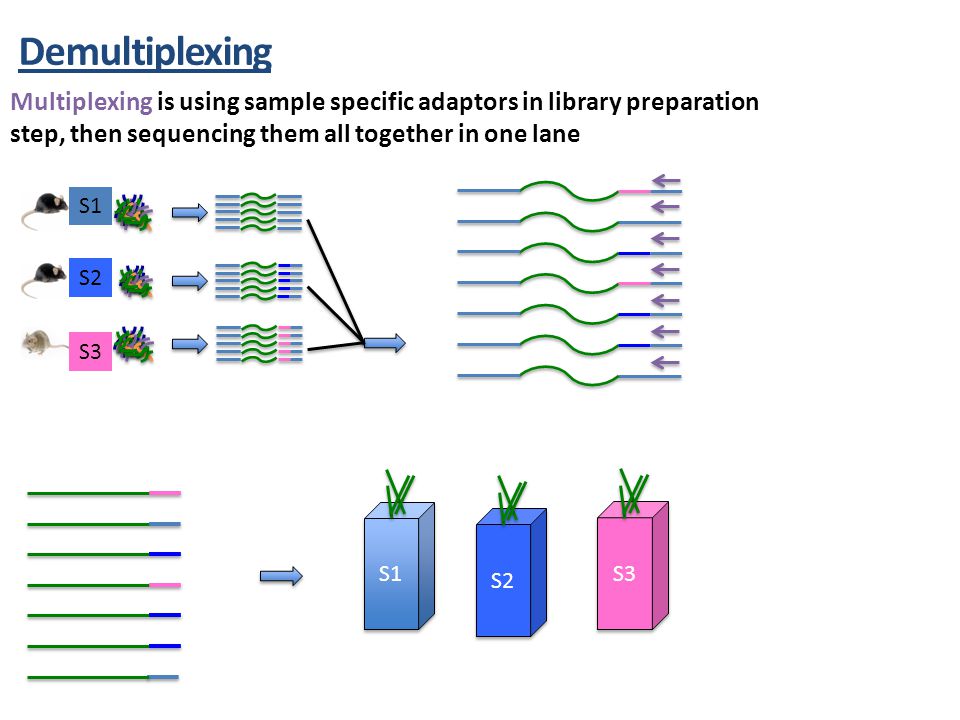
Source: Martha Park - Institute for Quantitative & Computational Biosciences Workshop
- QIIME2 must import all data files and convert them
- They become custom QIIME2 format files
- Allows for compression to save space and allow faster access
- Importing sequences has lots of options
- Some sequencing centers may demultiplex into fastq file for each sample
- Single versus paired end sequencing
- Quality scores or not (if you generated the data you should have these)
- Barcodes may be in a separate file
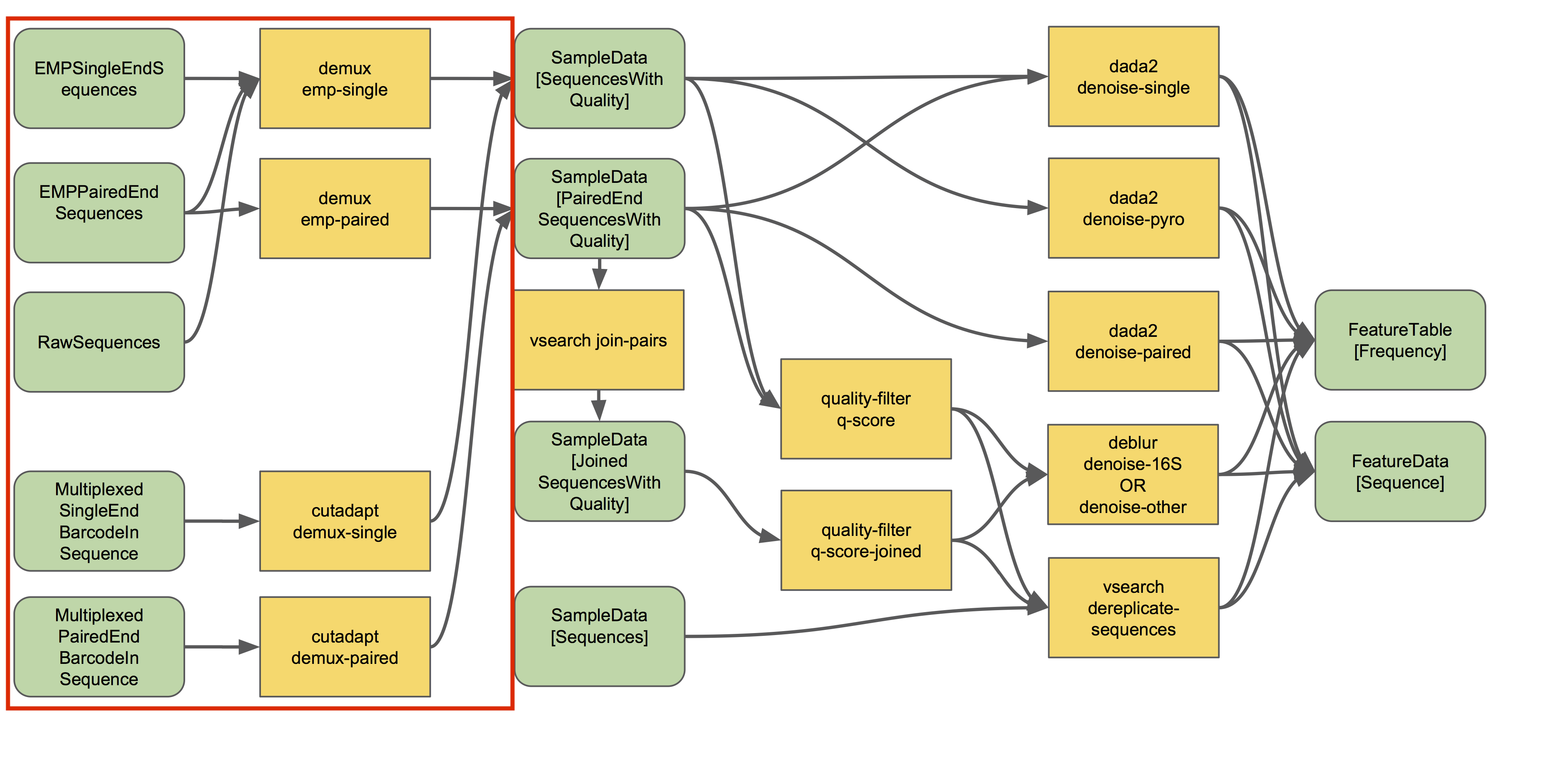
Source: QIIME2 - https://docs.qiime2.org/2018.8/tutorials/overview/#demultiplexing
Generally importing sequences will follow this format:
qiime tools import \
--type XXX \ # The format of your sequence files
--input-path XXX \ # The folder or manifest file where sequences are located
--input-format XXX \ # Tell QIIME2 how to import the sequences properly
--output-path XXX # Where the processed sequences should be saved
Fastq Manifest Format
This is the most flexible approach if the sequencing center demultiplexed your samples
- Make a manifest telling QIIME2:
- Which files align to which samples
- The paths to each of those files
- The direction: forward or reverse
- Manifest Format
- CSV - comma separated values in each column
- sample-id must match the sample names in your mapping file
- Header line must match below
- Save as CSV file
sample-id,absolute-filepath,direction #header line
sample-1,$PWD/some/filepath/sample1_R1.fastq,forward
sample-1,$PWD/some/filepath/sample1_R2.fastq,reverse
- Phred 33 vs 64
- Phred scores are sometimes offset by 31, depends on sequencer software
- This is important because they are always converted to 33 - Ask sequencing center!
- If you can ask for 33, as the 64 can be slow to convert for large files
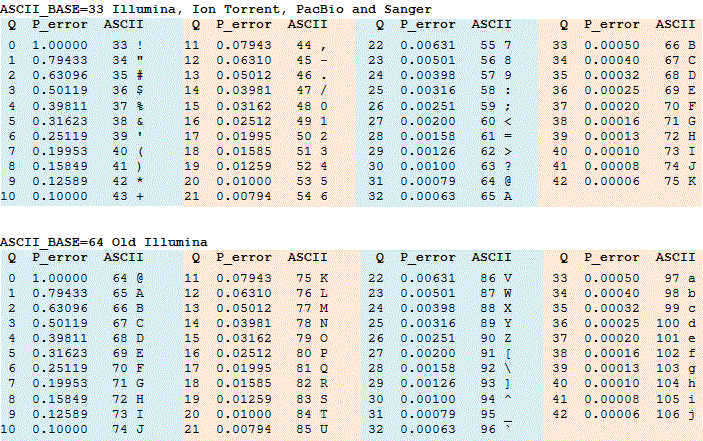
Phred score encoding for 64 and 33. Source: https://www.drive5.com/usearch/manual/quality_score.html
- Command options:
-input-format - SingleEndFastqManifestPhred33
- SingleEndFastqManifestPhred64
- PairedEndFastqManifestPhred33
- PairedEndFastqManifestPhred64
-type - ‘SampleData[SequencesWithQuality]’
- ‘SampleData[PairedEndSequencesWithQuality]’
-input-path - Path to your manifest file or folder of files
-output-path - Where QIIME2 will save the processed sequences in .qza format
Depending on your formats it will look something like:
# Example for Paired end with Phred 64
qiime tools import \
--type 'SampleData[PairedEndSequencesWithQuality]' \ # Change for paired/single end sequences
--input-path my_manifest.txt \
--output-path 1_0_input_seqs.qza \
--input-format PairedEndFastqManifestPhred64 # Change for paired/single and phred format
Earth Microbiome Project format
This is a more specific format if you follow the EMP protocols and the sequencing center does as well
A good approach if your sequences are not demultiplexed (all samples sequences together)
- All files in fastq format - often .gz indicates GunZip compression
- Single End:
- Fastq file of sequences - sequences.fastq.gz
- Fastq file of separated barcodes (by sequencing center) - barcodes.fastq.gz
- Paired End:
- Fastq file of forward sequences - forward.fastq.gz
- Fastq file of reverse sequences - reverse.fastq.gz
- Fastq file of separated barcodes (by sequencing center) - barcodes.fastq.gz
Place these files in a folder (my_seqs/), and use that folder name as the –input-path argument:
# Single End
qiime tools import \
--type EMPSingleEndSequences \
--input-path my_seqs \
--output-path 1_0_input_seqs.qza
# Paired End
qiime tools import \
--type EMPPairedEndSequences \
--input-path my_seqs \
--output-path 1_0_input_seqs.qza
There are other options for inputting sequences, check the QIIME2 documentation if these don’t fit your data
Importing, Demultiplexing, and Sequence Quality Control¶
Let’s try it out with some example data!

Source: giphy.com
First lets take a look at the mapping file to understand how QIIME2 creates visuals
qiime metadata tabulate \
--m-input-file paired_end/metadata.tsv \
--o-visualization paired_end/1_0_metadata_stats
- This will output a file 1_0_metadata_stats.qzv
- .qzv files are QIIME2’s visualization files
- They can be opened with ‘qiime tools view …’
- .qza files are QIIME2’s data files
- These cannot be viewed, and are often compressed
- Other scripts can often create visual .qzv files from .qza
Let’s see what we made
qiime tools view paired_end/1_0_metadata_stats.qzv
To import sequences and demultiplex…
- You should navigate to the folder called data/
- We will use the folder paired_end - This contains multiplexed sequence files and a metadata file 1. Forward sequences - reverse.fastq.gz 2. Reverse sequences - forward.fastq.gz 3. Barcode sequences - barcodes.fastq.gz 4. Metadata file - metadata.tsv
- We will import the sequences into QIIME2 format
- We will demultiplex the sequences
- We will examine the distribution of sequences across each sample
# See the files
ls -lsh paired_end/raw_seqs/
# Import the files into QIIME2 format
qiime tools import \
--type EMPPairedEndSequences \
--input-path paired_end/raw_seqs/ \
--output-path paired_end/1_0_input_seqs.qza
# See the new output file
ls -lsh paired_end
# Demultiplex the sequences based on barcodes in mapping file
qiime demux emp-paired \
--m-barcodes-file paired_end/metadata.tsv \
--m-barcodes-column BarcodeSequence \
--i-seqs paired_end/1_0_input_seqs.qza \
--o-per-sample-sequences paired_end/1_1_demultiplexed_seqs \
--p-rev-comp-mapping-barcodes
# See the new output file
ls -lsh paired_end
# Summarize the sequences per sample
qiime demux summarize \
--i-data paired_end/1_1_demultiplexed_seqs.qza \
--o-visualization paired_end/1_2_demultiplexed_seqs_summary.qzv
# Open the summary
qiime tools view paired_end/1_2_demultiplexed_seqs_summary.qzv
Click the ‘Interactive Quality Plot’ tab to view Phred scores
QIIME2 has our sequences… now what?
- If they are paired end then we need to join them
- Aligns forward and reverse sequences
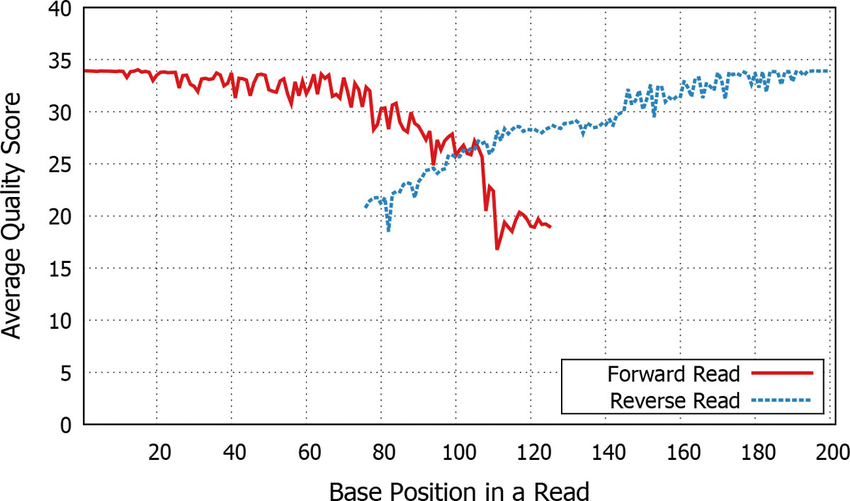
Paired end quality scores across sequence. Source: Kwon, S. Lee, B. Yoon, S. doi: 10.1186/1471-2105-15-S9-S10
Let’s align the paired end sequences and then look at some summary info
##### JOIN PAIRED ENDS #####
qiime vsearch join-pairs \
--i-demultiplexed-seqs paired_end/1_1_demultiplexed_seqs.qza \
--o-joined-sequences paired_end/1_3_joined_seqs.qza
# See the new output file
ls -lsh paired_end
##### SUMMARIZE JOINED SEQUENCES #####
qiime demux summarize \
--i-data paired_end/1_3_joined_seqs.qza \
--o-visualization paired_end/1_4_joined_summary
# VIEW THE RESULTS IN BROWSER #
qiime tools view paired_end/1_4_joined_summary.qzv
- Note how input and output files are named incrementally!
- This keeps them in order when you open in a folder
- Allows logic flow of code and files to align
- Keeps everything organized when you may generate hundreds of files!!!
There are other ways to join reads, here are some other read joining options.
** Finally we should filter low quality sequences with Phred scores**
##### PERFORM QUALITY CONTROL BASED ON PHRED Q SCORES #####
qiime quality-filter q-score-joined \
--i-demux paired_end/1_3_joined_seqs.qza \
--o-filtered-sequences paired_end/1_5_qc_seqs.qza \
--o-filter-stats paired_end/1_6_qc_seqs_summary.qza
#### SUMMARIZE JOINED SEQUENCES #####
qiime demux summarize \
--i-data paired_end/1_5_qc_seqs.qza \
--o-visualization paired_end/1_7_qc_summary
# VIEW THE RESULTS IN BROWSER #
qiime tools view paired_end/1_7_qc_summary.qzv
Keep in mind there are often more options than just the ones I am showing here…
To view all of the parameters available in a QIIME2 script follow just the function call with –help
# LIST ALL PARAMETERS FOR quality-filter q-score-joined SCRIPT #
qiime quality-filter q-score-joined --help
For example here are all of the useful options for filtering by quality scores
--i-demux ARTIFACT_PATH The demultiplexed sequence data to be quality filtered. [required] --p-min-quality INTEGER The minimum acceptable PHRED score. All PHRED scores less that this value are considered to be low PHRED scores. [default: 4] --p-quality-window INTEGER The maximum number of low PHRED scores that can be observed in direct succession before truncating a sequence read. [default: 3] --p-min-length-fraction FLOAT The minimum length that a sequence read can be following truncation and still be retained. This length should be provided as a fraction of the input sequence length. [default: 0.75] --p-max-ambiguous INTEGER The maximum number of ambiguous (i.e., N) base calls. This is applied after trimming sequences based on min_length_fraction. [default: 0] --o-filtered-sequences ARTIFACT_PATH SampleData[JoinedSequencesWithQuality] The resulting quality-filtered sequences. [required if not passing –output-dir] --o-filter-stats ARTIFACT_PATH Summary statistics of the filtering process. [required if not passing –output-dir] --output-dir DIRECTORY Output unspecified results to a directory
Deblur - The New OTU¶

Traditional Operational Taxonomic Unit (OTU) clustering
How do we actually charicterize a community?
Lump similar sequences together - There are a variety of algorithms
- OTUs - Sequence similarity % cutoff
- Deblur - Identical sequences based on likelihood of being biological versus artifactual noise
- DADA2 - Similar sequences based on likelihood of being biological versus artifactual noise
Assign taxonomy to each of the sequence clusters or OTUs - Done using reference databases of known taxon for the amplicon of interest
Total the counts for each cluster across each sample into observation table
We will use Deblur. DADA2 is now really the other option (don’t use traditional % cutoffs).
There are advantages and caveats to each, but they get very similar results for the most part.
- To get Deblur ‘features’ we will:
- Evaluate the quality (Phred) scores along the sequences
- Trim the sequences to the same length
- Use Deblur to ‘denoise’ - remove likely sequencing errors
- Use Deblur to count the remaining sequences for each feature
First let’s decide where to trim our sequences… click the ‘Interactive Quality Plot’ tab
##### INVESTIGATE THE QUALITY SCORE PLOT TO PICK TRIM LENGTHS BELOW #####
qiime tools view paired_end/1_7_qc_summary.qzv
Note in the middle how the paired end overlap leads to higher Phred confidence!
Let’s trim to 240bp just before the dip…
This is somewhat arbitrary, but generally pick a point where the quality drops and make sure you change in your script!
There are many options for the next step, think about your situation
Deblur! Lets denoise our sequences into a feature table
##### DEBLUR DENOISE DUPLICATE SEQUENCES AND CREATE OBSERVATIONS #####
qiime deblur denoise-16S \
--i-demultiplexed-seqs paired_end/1_5_qc_seqs.qza \
--p-trim-length 240 \
--o-representative-sequences paired_end/2_0_deblur_representative_seqs.qza \
--o-table paired_end/2_1_deblur_table.qza \
--p-sample-stats \
--o-stats paired_end/2_2_deblur_stats.qza
# See output files
ls -lsh paired_end
Deblur can take a while to run, for our purposes you can just move the output files from the extras folder
To cancel the run press ctrl-c
# Remove any files if generated
rm paired_end/2_*
# See current files
ls -lsh paired_end
# Move the files
cp paired_end/extras/* paired_end/
# See output files
ls -lsh paired_end
Now we have generated:
- Representative sequences: 2_0_deblur_representative_seqs.qza
- These will be used for alignment, phylogeny building, and taxonomic assignment
- Observation table: 2_1_deblur_table.qza
- These are the counts for each representative sequence across each sample
- Statistics: 2_2_deblur_stats.qza
- Allows us to examine counts across samples
Let’s look at the stats…
##### DEBLUR VISUALIZE STATISTICS #####
qiime deblur visualize-stats \
--i-deblur-stats paired_end/2_2_deblur_stats.qza \
--o-visualization paired_end/2_3_deblur_stats_summary.qzv
qiime tools view paired_end/2_3_deblur_stats_summary.qzv
And summarize the table…
##### DEBLUR SUMMARIZE TABLE #####
qiime feature-table summarize \
--i-table paired_end/2_1_deblur_table.qza \
--o-visualization paired_end/2_4_deblur_table_summary.qzv \
--m-sample-metadata-file paired_end/metadata.tsv
qiime tools view paired_end/2_4_deblur_table_summary.qzv
And look at the representative sequences…
##### DEBLUR SUMMARIZE SEQUENCES #####
qiime feature-table tabulate-seqs \
--i-data paired_end/2_0_deblur_representative_seqs.qza \
--o-visualization paired_end/2_5_deblur_representative_seqs_summary.qzv
qiime tools view paired_end/2_5_deblur_representative_seqs_summary.qzv
Neat! Well done!
Alignment into Amplicon Phylogeny¶
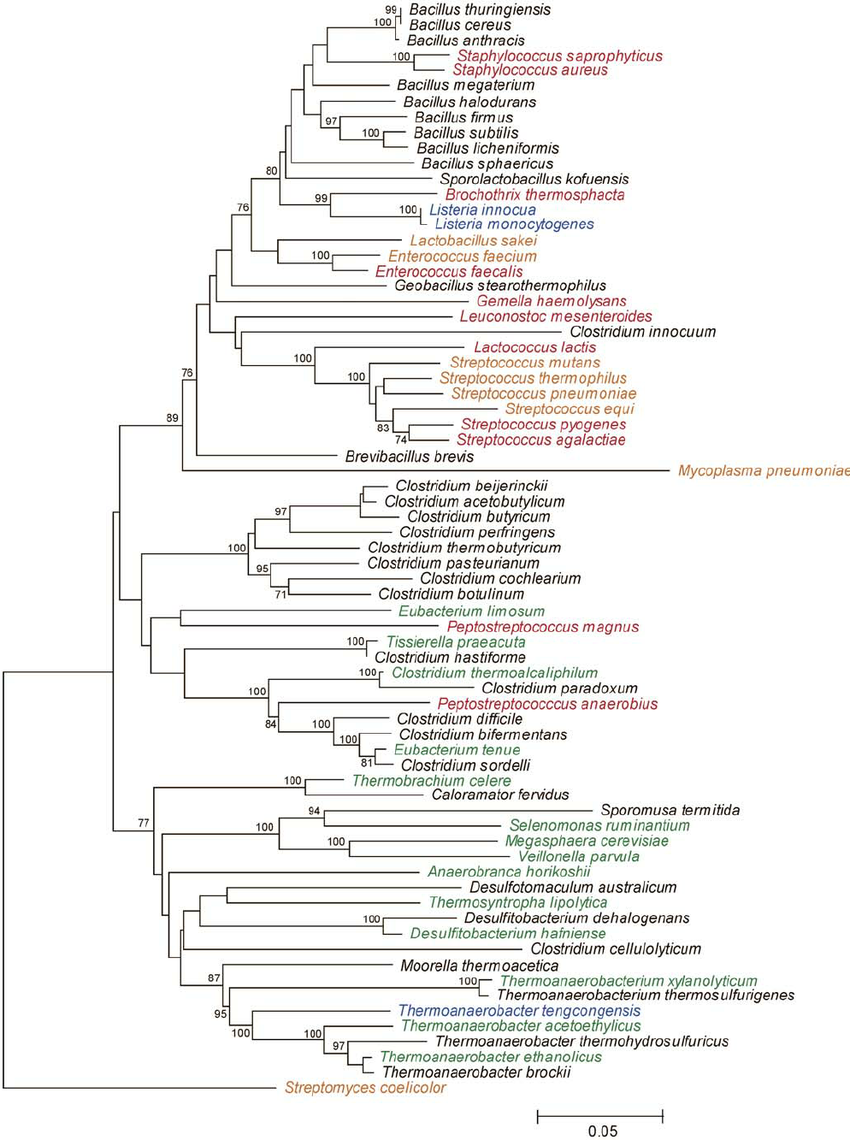
Example phylogeny. Source: Rob Onyenwoke et. al. doi: 10.1007/s00203-004-0696-y
- A phylogeny depicts the genetic relatedness between each of our amplicons
- Used for diversity metrics incorporating genetic relatedness of features
- To construct a phylogeny of our representative sequences we will..
- Align the sequences using Mafft
- Mask highly variable positions in the alignment (removes uninformative noise)
- Construct an unrooted phylogeny with FastTree
- Root the phylogeny at the tree’s midpoint
##### CREATE ALIGNMENT OF REPRESENTATIVE SEQUENCES #####
qiime alignment mafft \
--i-sequences paired_end/2_0_deblur_representative_seqs.qza \
--o-alignment paired_end/3_0_deblur_aligned_seqs.qza
##### MASK HIGHLY VARIABLE NOISY POSITIONS IN ALIGNMENT #####
qiime alignment mask \
--i-alignment paired_end/3_0_deblur_aligned_seqs.qza \
--o-masked-alignment paired_end/3_1_deblur_aligned_masked.qza
##### CREATE PHYLOGENY WITH FASTTREE #####
qiime phylogeny fasttree \
--i-alignment paired_end/3_1_deblur_aligned_masked.qza \
--o-tree paired_end/3_2_deblur_tree_unrooted.qza
##### ROOT PHYLOGENY AT MIDPOINT #####
qiime phylogeny midpoint-root \
--i-tree paired_end/3_2_deblur_tree_unrooted.qza \
--o-rooted-tree paired_end/3_3_deblur_tree.qza
# look at files
ls -lsh paired_end
Taxonomy Assignment¶
- Finally before we can start analysis we want to assign taxonomy to each of our representative sequences
- More biologically informative than sequences with arbitrary ids
- Allows us to bin by known classifications
- We will use the GreenGenes 94% reference database for a minimal example, but I would suggest another database for your actual project
- Silva
- GreenGenes 99%
- Other options, please learn which is best for your project!
- **First import the GreenGenes Database*
- 94% Unique 16S Sequences
- Taxonomy Reference Table
- Contains number of ribosomal DNA copies for each taxon
##### ASSIGN TAXONOMY #####
# !!! SET PATH TO TAXONOMY REFERENCE !!! #
greengenes_path=greengenes/
### IMPORT TAXONOMY REFERENCE SEQUENCES ###
qiime tools import \
--type 'FeatureData[Sequence]' \
--input-path "$greengenes_path"94_otus.fasta \
--output-path paired_end/greengenes_94_otus.qza
### IMPORT TAXONOMY REFERENCE TABLE ###
qiime tools import \
--type 'FeatureData[Taxonomy]' \
--input-format HeaderlessTSVTaxonomyFormat \
--input-path "$greengenes_path"94_otu_taxonomy.txt \
--output-path paired_end/greengenes_94_taxonomy.qza
- Next we will:
- Train a taxonomy classifier for our amplicon region in the reference database
- Naive Bayes Classifier
- We will identify our region of interestusing the primer seqeunces
- Select only region of the total 16S that was amplified and sequenced
- Apply the classifier to our 16S Amplicon Sequences
- Each sequence is queried and taxonomy assigned to the closest confident level
### EXTRACT REFERENCE READS BASED ON PRIMERS AND TRIM LENGTH ###
qiime feature-classifier extract-reads \
--i-sequences paired_end/greengenes_94_otus.qza \
--p-f-primer AGAGTTTGATCCTGGCTCAG \
--p-r-primer GCTGCCTCCCGTAGGAGT \
--p-trunc-len 320 \
--o-reads paired_end/greengenes_94_seqs_extracted.qza
### TRAIN NAIVE BAYES CLASSIFIER FOR TAXONOMY ASSIGNMENT ###
qiime feature-classifier fit-classifier-naive-bayes \
--i-reference-reads paired_end/greengenes_94_seqs_extracted.qza \
--i-reference-taxonomy paired_end/greengenes_94_taxonomy.qza \
--o-classifier paired_end/greengenes_94_classifier.qza
### ASSIGN TAXONOMY WITH TRAINED CLASSIFIER ###
qiime feature-classifier classify-sklearn \
--i-classifier paired_end/greengenes_94_classifier.qza \
--i-reads paired_end/2_0_deblur_representative_seqs.qza \
--o-classification paired_end/3_4_assigned_greengenes_94_taxonomy.qza
Finally let’s visualize our taxonomic assignment
### MAKE TAXONOMY OUTPUT VISUALIZATION ###
qiime metadata tabulate \
--m-input-file paired_end/3_4_assigned_greengenes_94_taxonomy.qza \
--o-visualization paired_end/3_5_taxonomy_visualization.qzv
qiime tools view paired_end/3_5_taxonomy_visualization.qzv