Experimental Design and Sample Preparation¶
Key Takeaways
- There are caveats and biases in every method
- Understand the biases in your methods
- Understand how that bias will affect results
Plan and optimize well, then stick to one protocol!
Experimental Design¶
How do I start a microbiome amplicon study? Plan ahead!
It is particularly important to consider the details of your study before you start
- Cannot add samples later
- Cannot remove contamination after samples are collected
- Cannot add controls necessary to validate quality after sequencing
- Cannot remove bias if other factors very with treatment of interest
- Cannot get more microbial DNA if host DNA dominates

Source: Aaron Bacall Source: www.art.com
- Start with a question and hypothesis.
- Consider expected effect size on microbiome composition
- Calculate how many samples will be needed to detect if the effect exists
- Consider changes community wide and detecting changes in taxon abundance and presence
- Keep in mind not every sample will pass every QC step - samples will be lost
- Larger N
- Parallel processing replicates
- Always step back and ask: “Will a whole community profile answer my question?”
Often people hypothesize that “the microbiome will change”, without considering what that actually means - Amplicon sequencing doesn’t tell you community function - It won’t give you strain or even consistent species level resoultion
- In addition to E. coli, there are five other species in the genus Escheria and some are commensal in the human gut
Sampling and Controls¶
How will you collect your samples?
Generally consider in collection:
- Size of sample
- Determines available DNA
- Can you control an equal amount from each subject - absolute quantification
- Reserve sample - mix evenly so reserve is the same as utilized sample
- Sampling vessel
- Ease of collection
- Sterility
- Stabilization solution
- Need DNA and RNA
- Other ‘omics - i.e. mass spectrometry affected by salt concentration
Source: www.dnagenotek.com
- Storage
- Immediate storage -80C
- Long term stabilization solutions can introduce bias
- Prevent bacterial overgrowth at room temperature
Key controls to keep in mind:
Uniformity
- Collect samples at the same time and method across treatment groups
- Same for processing DNA extraction and library preparation for sequencing
Randomization
- Consider many factors that could vary across treatment groups
- Time of day, season
- Sampling in same location
- Mouse cage effects
- Co-housing
- Mixing bedding and food
Sterility
- Collect samples, but also sample microbes in the local environment if possible
- Always include blank water samples
- Collection blank
- Extraction blank
- Library preparation blank
- Sequencing blank
- Work in biosaftey hood whenever possible
- Particularly sensitive to contamination before PCR steps
- Mock community
- Known taxonomy and abundance
- Generous donor
- If sampling / extracting / sequencing in batches include one consistent sample across each
If working with human subjects:

Source: Charis Tsevis
- Consider ease of self-collection
- Different body sites have very different communities
- Even location on a stool sample (inside versus outside)
- Biopsies are loaded with human DNA
- IRB compliant methods
- Gloves
- Way to catch stool in toilet and disposal
- Proper instructions
- Immediate acquisition versus returning samples later
- Cannot control amount provided if self-collection
- Must stablize to prevent bacterial overgrowth, or ‘blooms’
Source: www.dnagenotek.com
DNA Extraction¶
DNA extraction techniques vary significantly in community bias
- Every kit introduces bias, so pick one and stick with it!
- There are many options, so research which is best for your long term goals
- The Earth Microbiome Project has well documented protocols.
- Efficiency at lysing cells is important
- Bead beating versus chemical lysis
- Mechanical bead beating seems to be most thorough gram +/-
- Keep in mind bead beating heats the sample through friction, don’t over-do it!
- Take time to properly optimize and document
- Always note extraction batch, who performed the extraction, and the kit lot number
- Always include extraction blanks, all kits have contaminants - kit’ome.
- Some kits perform dual DNA and RNA extraction
Amplification¶
PCR amplification of the marker gene of interest
- Use known protocols and established primers
- Different variable regions bias the community differently, stick to what’s known!
- The Earth Microbiome Project has well documented PCR protocols (16S, 18S, ITS)
- Always optimize your PCR for the expected sequence length and concentration
- Include PCR blank to control for processing contamination
- Sequencing Depth
- Generally 10k - 100k sequences per sample is adequate coverage
- Diminishing returns at greater depths
- Taxonomic resolution based on sequence length not sequencing depth
- Many reads increase sequencing errors, qc filtered
- Analyses are rarefied or relative abundance
- Rare organisms will still be rare, depth doesn’t change proportions
Earth Microbiome Project V4 Primers
515F FWD: GTGYCAGCMGCCGCGGTAA
806R REV: GGACTACNVGGGTWTCTAAT
The primer sequences in EMP protocols are always listed in the 5′ -> 3′ orientation. This is the orientation that should be used for ordering.
Components of full reaction:
_____5′ Illumina adapter_________________________Golay barcode_____Pad________Linker___Forward primer
FWD: AATGATACGGCGACCACCGAGATCTACACGCT XXXXXXXXXXXX TATGGTAATT GT GTGYCAGCMGCCGCGGTAA
- Each sample PCR reaction will be performed with it’s own unique barcode combination
- This allows bioinformatic demultiplexing - assigning sequences to their respective sample
Sequencing Platforms¶
Illumina
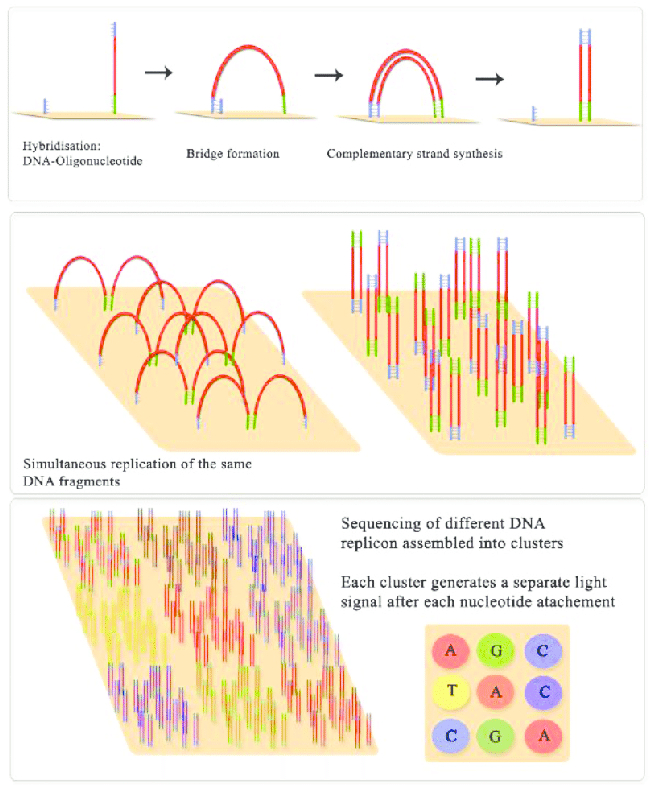
Illumina sequencing molecular biology Source: Jaroslaw Grzadziel (Research Gate)
- By far the most widely used sequencing method
- Millions of sequences allow multiplexing many samples on one run
- High coverage of the community (~10k sequences per sample is ideal depending on complexity)
- Short sequences 100-250bp generally cover 1-3 variable regions
- Allows high coverage, but lose resolution past genus level
- Paired end is better than single end
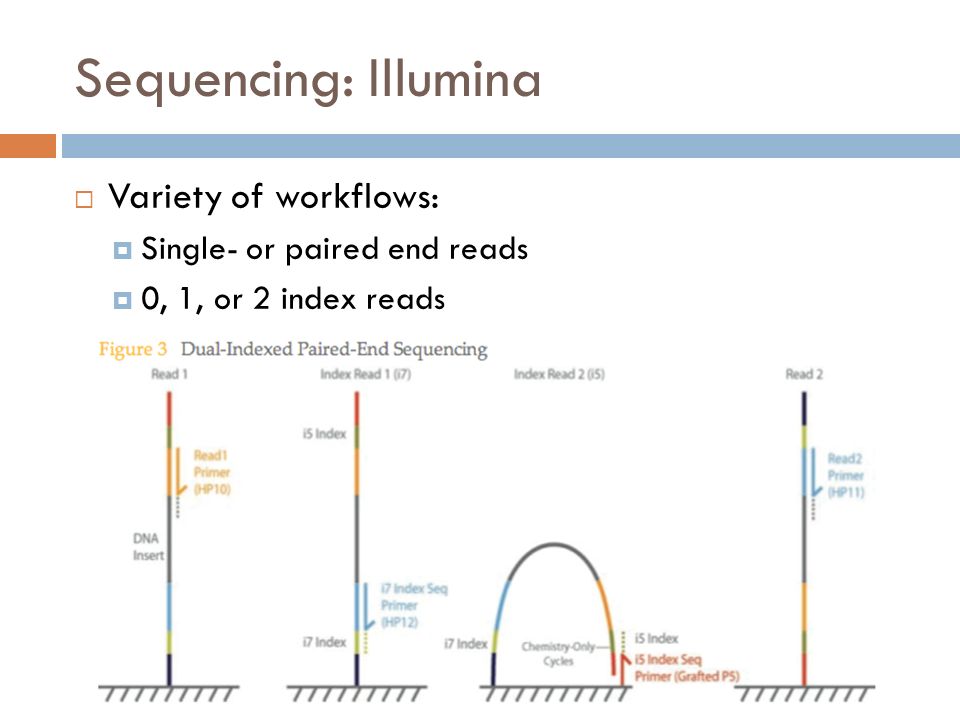
Mechanism of paired end Illumina sequencing Source: Christine King
- Illumina Amplicon Sequencing at VANTAGE
- 250 bp sequence reads (Covers entire V4 region)
- Dual-index sequencing strategy - increased multiplexing
- Each MiSeq run produces >5,000,000 reads (Great for multiplexing)
- Best option for 16S rRNA gene amplicon sequencing
- VANTAGE also offers HiSeq, NextSeq, and NovaSeq Illumina sequencing platforms
- MiSeq has 250bp length, other platforms only offer shorter reads so plan ahead
PacBio and Nanopore
- Could do full 16S gene
- Very high strain level resolution
- But much lower coverage and multiplexing = higher costs
- Higher error rates
Mapping File¶
How do you store all of this information about samples in a useful way?
- A mapping file is composed of:
- Each row representing a sample
- Each column representing some information about each sample
- Are TSV - tab separated values
- TSV is a .txt output format from Excel
- A header line starting with a # indicates the column names
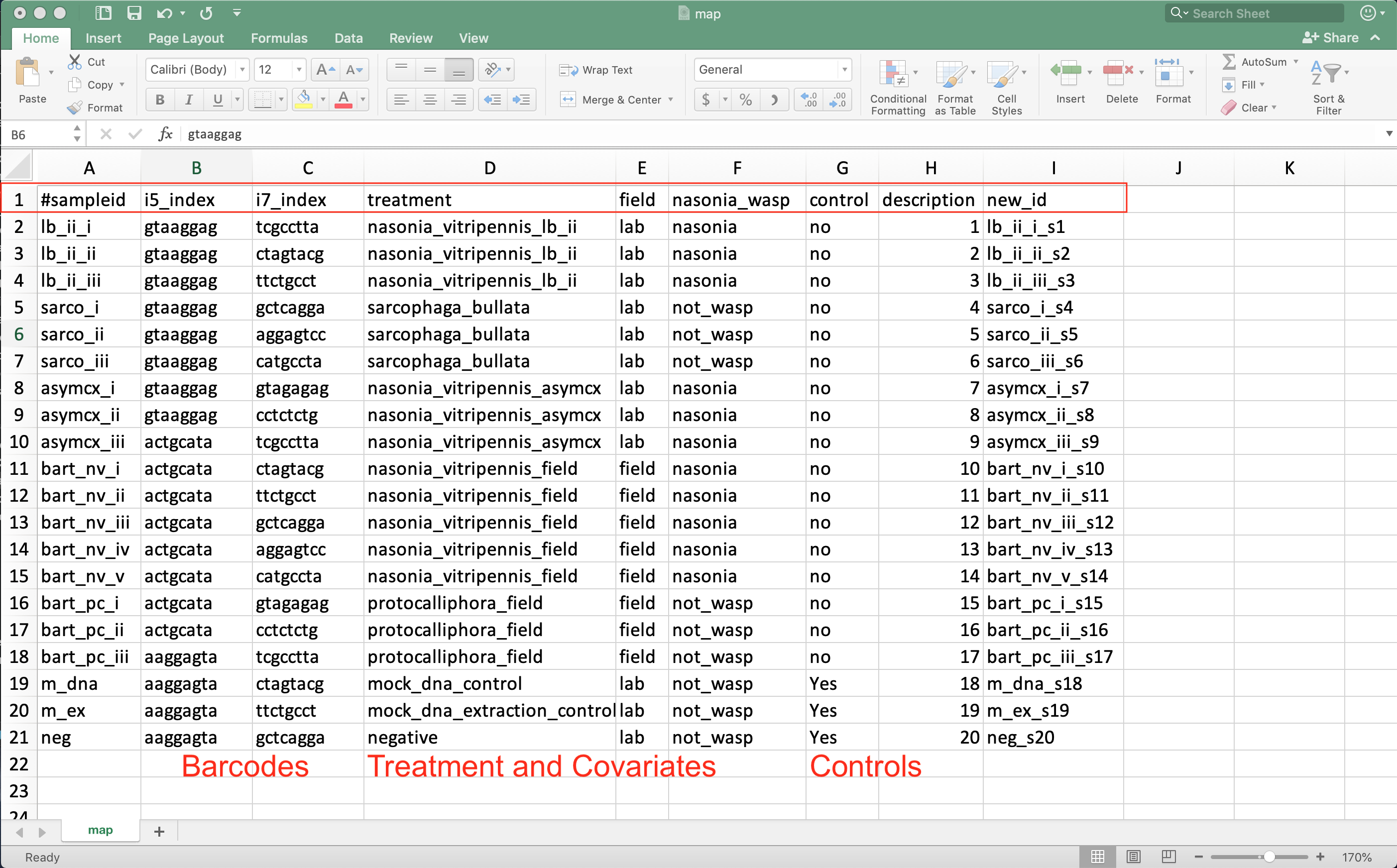
Source: Example mapping file in excel.
- Always start with #sampleid column
- A unique id for each sample - cannot be the same as any other sample
- Include columns for the unique sample barcodes
- Allows demultiplexing of sequences to their respective sample
- Also include control and qc information about DNA extraction batch, person extracting, PCR batch, sequencing run, cage…
- Allows testing if these factors influenced the community composition
- Have columns indicating which samples are blanks, mock community, and generous donor samples
- Have columns for your treatement and any covariates of interest
- Age, sex, BMI, collection season…
- Last column should be description if you need backward compatibility with QIIME1
For more detailed information check out the full QIIME2 Metadata Guide!
Best Practices
- Rule Number 1: Don’t get fancy!
- Don’t use spaces, use _ (underscore) instead
- Except #sampleid, the only punctuation in this column can be dashes ‘-‘ (no underscores)
- Don’t include other types of punctuation, this will only cause problems later on!
- Leading and trailing white spaces are ignored
- I’d stick to all lower-case (case insensitive) characters
- Not required, but may save you a lot of trouble with weird errors later on!