Microbiome Introduction¶

Source: http://microbeminded.com/
A biome is a complex ecosystem of biological organisms, often defined and shaped by a shared ecosystem.
Therefore, a microbiome is a complex ecosystem of microorganisms:
- Bacteria
- Eukaryotic Fungi and Protists
- Archaea
- Viruses and Phages
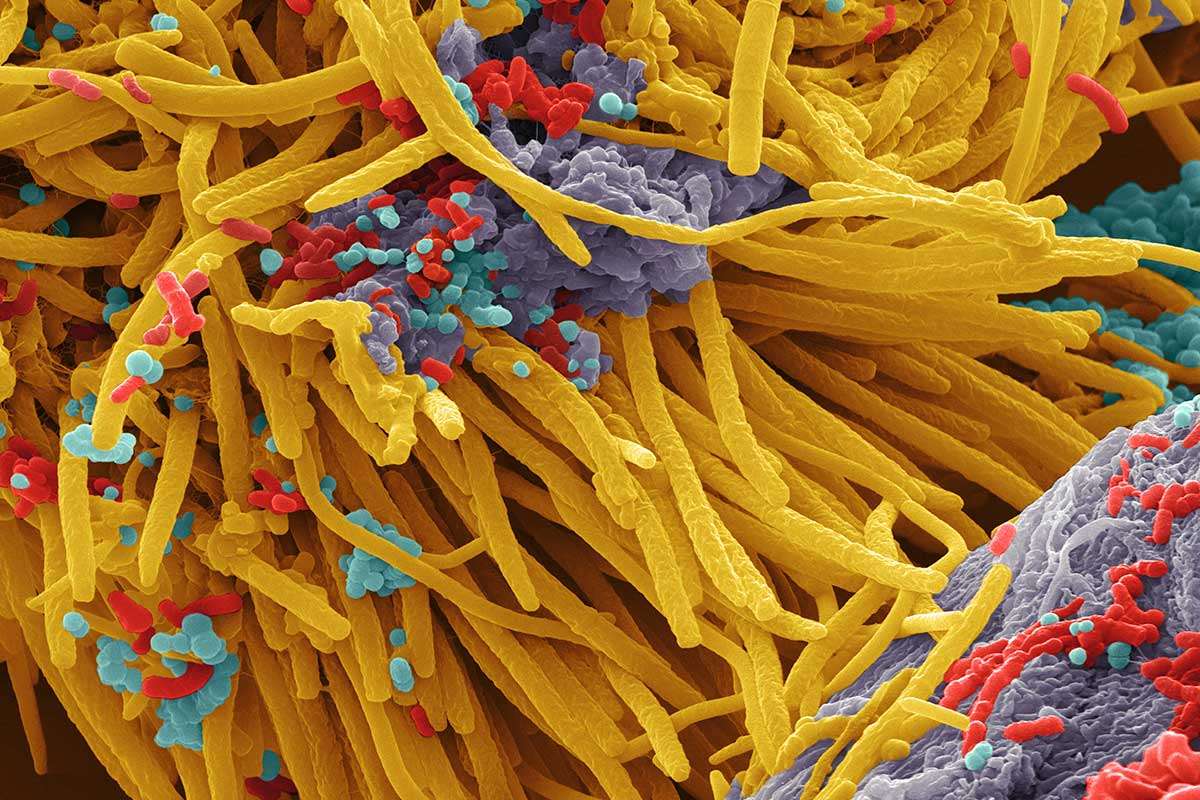
Scanning electron image of a complex microbial community. Colors are artificial. Source: https://www.newscientist.com
There are very few places on earth where microbiomes do not exist
- Deep sea vents
- A mile underground
- High Atmosphere
- Middle of the ocean
- On and inside pretty much every animal and plant (outside of the lab)
Since animals and eventually humans arose they have been ubiquitously interacting with and evolving alongside microbial communities
Definitions¶
In popular science and among the public the term microbiome is thrown around as a community of microorganisms.
Within the research community however there is more nuance (but microbiome is still used loosely), so let’s clearly define some important terms:
- Microbiota - The microorganisms forming a complex community, often charicterized using taxonomy.
- The bacterial component is what we will examine with 16S Amplicon Sequencing.
- Who is there?
- Microbiome - The total genomic composition of all organisms in a microbiota.
- Examined with Metagenomic Shotgun Sequencing. Not restricted to bacteria.
- What could they do?
- Metagenome - The microbiome as captured with high-throughput genetic sequencing.
- Captures random sections of DNA from all microorganisms and macroorganisms.
- What functions are available to the community?
- Metatranscriptome - The expressed RNA components of a microbiome captured with high-throughput genetic sequencing.
- RNA is extracted, reverse transcribed, and sequenced.
- What are they trying to do?
- Proteome - The protein profile resulting from transcription and translation of the microbiome.
- Often examined with mass spectrometry or sometimes NMR.
- What are the actively able to do?
- Metabolome - The profile of metabolites and biologically active chemicals.
- The metabolome is shaped by enzymatic activity of the proteome.
- Most often examined with targeted or untargeted mass spectrometry.
- How do their actions shape the abiotic environment?

Like a nesting doll family, there are many layers that can each tell their own part of the story. Source: Giphy.communities
The Human Microbiome¶

What are the biomes of microbes on and inside the human body? Source: http://microbeminded.com/
It is estimated that there are roughly the same number of human and microbial cells in your body!
- This encompases trillions of living microorganisms.
- Surveys often report hundreds to thousands of unique bacterial species.
- Microbiome diversity of 10 million+ unique genes, outnumbering our genetic repertoire 500:1.
- Vast majority are along digestive tract, but skin and vagina have unique communities as well.
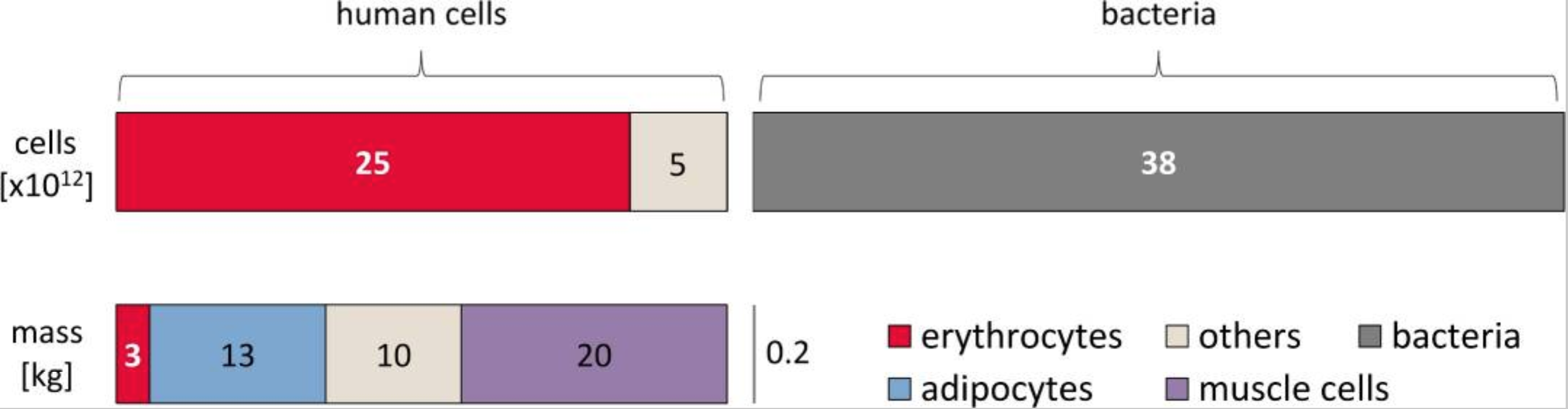
Estimating counts and mass of cells in the human body. Source: Sender, R. et al. PLoS Biology. 2016.
While highly abundant, microbes are much smaller than human cells.
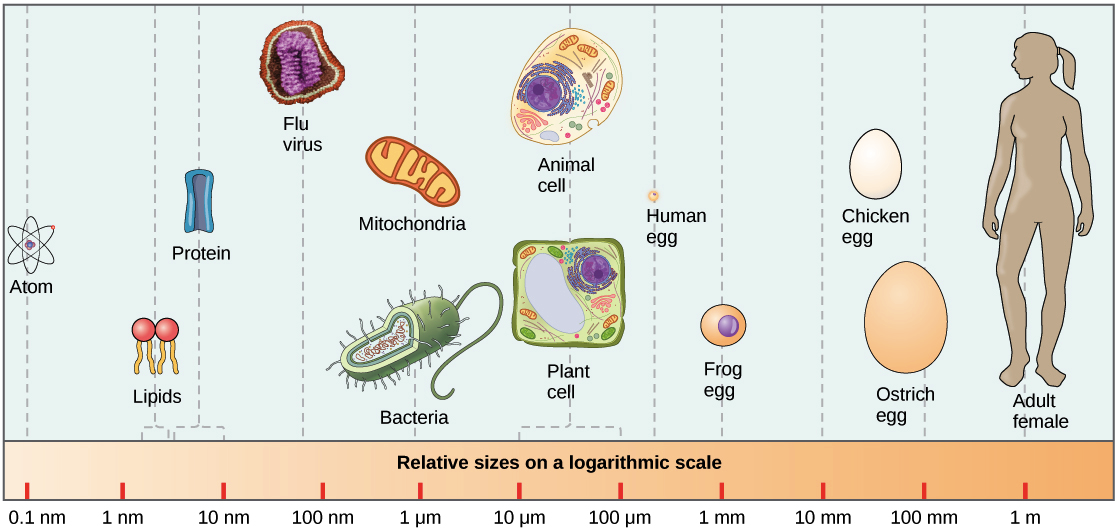
Most microbes are 10-100x smaller than human cells (same size as mitochondria). Source: https://courses.candelalearning.com
16S Ribosomal DNA and Phylogenetic Markers¶
So how can we look at which microbes make up a microbiota?
The methodology we are discussing today falls under the umbrella of Amplicon Sequencing.
In amplicon sequencing, one gene shared by all microorganisms of interest is amplified and sequenced to charicterize who is in the microbiota?
- Amplicon sequencing involves:
- Extracting all DNA from a microbial community.
- PCR amplifying a specific marker gene shared by the microbes of interest.
- Sequencing all of the PCR amplicons.
- Assigning the genetic sequences back to reference databases of known microbes.
- Compiling counts of the number of sequences representing each taxon.
This is most often done for bacterial communities with sections of the DNA coding region of 16S Ribosomal RNA.
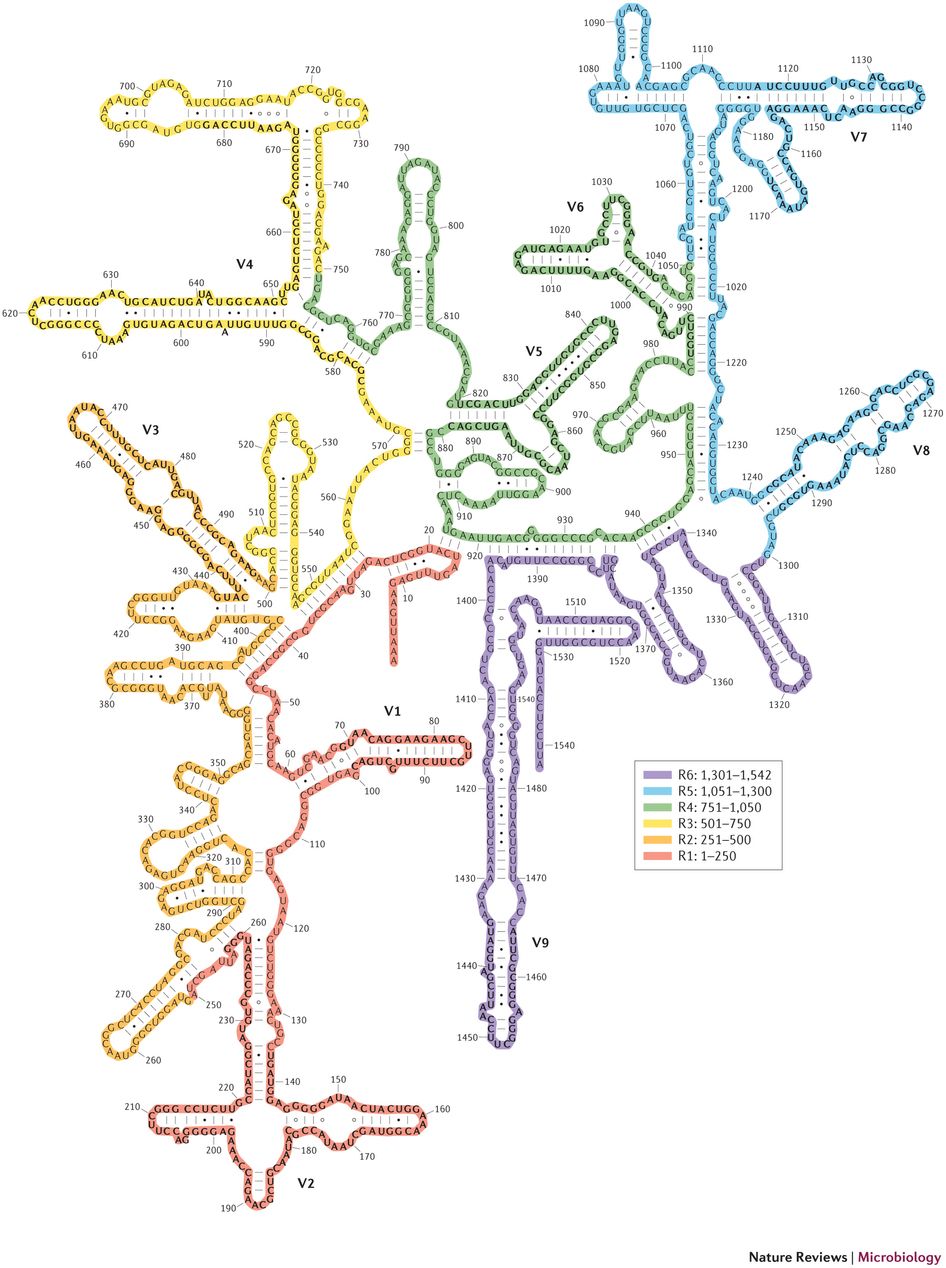
Secondary hairpin structure of 16S rRNA. Source: rna.ucsc.edu
16S rRNA Structure
- The RNA component of bacteria ribosomes forms bonds between nucleotide bases.
- The attraction of these bases forms secondary loop and hairpin structures.
- Tertiary structure forms and combines with protein components of the ribosome to form an active unit.
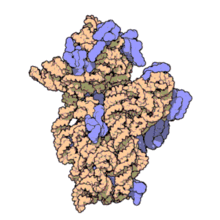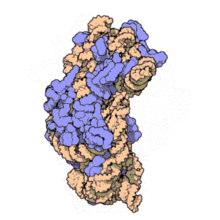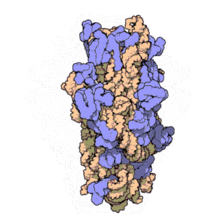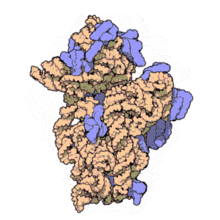
30S ribosome small subunit with 16S rRNA in orange and a number of purple proteins. Source: wikipedia.com
This is a useful gene because:
- It is shared by almost all bacteria and archaea.
- The ribosome is crucial to life
- RNA sequence is critical for tertiary structure backbone
- Leads to stabilizing selection
- For this reason it is slow evolving, particularly in regions important to the overall structure.
- Thus it has regions that are more conserved and regions that are more variable (evolutionary changes accrue more quickly).

Nine variable regions are each separated by highly conserved regions. Source: biology.stackexchange.com
16S rDNA is not the only marker gene used for amplicon sequencing, just the most common.
- Fungal communities are often targeted with the ITS (Internally Transcribed Spacer).
- Region between the 18S and 5.8S rRNA genes in Eukaryotes.
- Archaea are generally targeted with different 16S primers.
- They do not have enough homology with bacteria (captured with bacterial amplicons, but very biased).
- Bacteria usually vastly outnumber archaea, dominating sequencing power.
- I’ve heard of 18S and 28S being used for protists and fungi, but could really use many genes. Except…
Keep in mind before you try to design your own amplicons…
- Many genes are not under such strict conserving selection.
- Homology and even presence may be far more taxon restricted.
- Harder to target because of lack of conserved regions.
- PCR amplicons to a new gene or region require extensive validation.
- May not amplify different orthologs equally, thus biasing abundance.
- Could use mock community with known abundance as a control.
Therefore, don’t try to design your own primers unless you have a really good reason… and lots of time to validate.
- The main application would be interest in a smaller subset of taxa (i.e. betaproteobacteria class).
- Could provide better strain resolution, but I wouldn’t trust abundance.
- Allows more reads from organisms of interest, but miss everything else.
Advantages and Disadvantages¶
Key advantages of Amplicon Sequencing
There are very well studied protocols, amplicons, and tools.
- The Earth Microbiome Project protocol is the most widely accepted.
- Very well validated, and used in many studies including the Human Microbiome Project.
- Have protocols for 16S, 18S, and ITS detailing steps and best practices.
Generally the cheapest and most approachable method (compared to metagenomic sequencing as an alternative).
- Requires less sequencing depth per sample to get confident profile.
- This allows far more samples to be on a sequencing run.
- In 16S we usually want 10-100k assigned sequences per sample.
- In metagenomics 1-2 million sequences is the extreme low end, often want 10X that.
- Much more computationally approachable.
- Search against database of one gene vs all full genomes (including eukaryotic, host, viral…).
- Less likely to sequence any relevant amount of human DNA.
- Could make IRB approval easier.
- Don’t need to remove human DNA before publicly sharing, or having to restrict access to approved parties i.e. dbGAP.
- However, if there is very little microbial DNA I have seen mostly off target sequences from human mitochondria.
Key disadvantages of Amplicon Sequencing
- Amplicons can only tell you who is there and in what abundance.
- It cannot tell anything about the functional capabilities of the microbes.
- It is semi-quantitative at best, not absolute.
- Can really only examine relative abundance.
- Thus never know if one sample has more or less microbes, only a microbe’s ratio with other microbes.
- No idea if changes over time are because the total number of microbes increased, decreased, or just shifted members.
- Resolution is limited because the amplicons are short, so generally family or genera is the most that can be distinguished.
- Really cannot confidently distinguish species or strains within a genus.
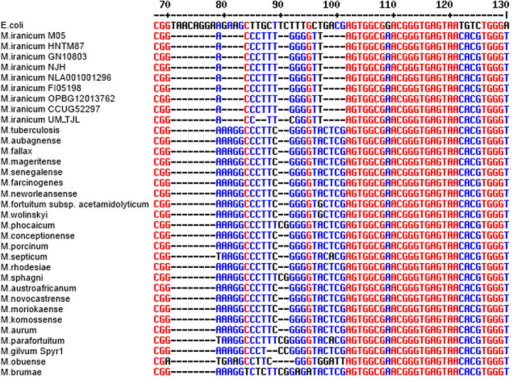
Aligning short reads means there may be very few differences to distinguish related taxa. Source: nature.com
- 16S rRNA gene copy number can vary by an order of magnitude between bacterial species.
- The databases try to correct for this, but it’s not perfect.
- This means there is some bias in the relative abundance of members.
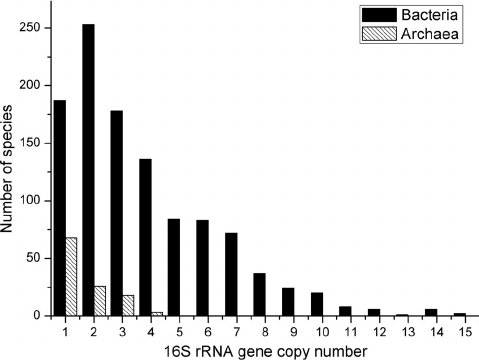
Most bacteria have two or more copies of rDNA in their genomes, which must be corrected for when counting totals. Source: researchgate.net
- Seeing DNA in a sample does not confirm that there are live microbes, just that their DNA was present.